Description
Features of Variant Ranker?
Getting started
Input format
Can I use multi sample VCF format?
Understanding the output
Examples
CaseControl Genotype filtering
Performance evaluation/Running times
Related Publication
Variant Ranker: is a webserver that performs ranking of variants in genomic datasets, integrates information from multiple sources to prioritize each variant by its deleteriousness, novelty and existing information. As there are several prediction/annotation tools available, there is often discrepancies amongst results. Variant Ranker aims to combine several prediction (deleteriousness, conservation) algorithms and annotation databases in order to provide the "optimised" ranked result
Features of Variant Ranker:- Rank (coding and non-coding variants), annotate and prioritise variants based on novelty, effect and existing information
- External links to UCSC geneome browser, ensembl and dbSNP
- Filtering Strategies: (Result Explorer)
- Functionally important variant filtering (filter across several databases including refGene, ensGene, clinVar, gwasCatalog, OMIM, 1000g, ENCODE elements etc )
- Case Control Allele Filtering
- Model of inheritance Filtering (dominant, recessive, X-Linked)
- Population frequency filtering (rare/common alleles)
- Downstream analysis (Network Analyser)
- Functional enrichment analsysis
- Exploratory network analysis
Variants are ranked according to the default set of weights which can also be adjusted by the user. Users can also select/deselect algorithms accordingly. The default set of weights (0-1) are pre-calculated according to functional importance; a higher weight is therefore given for a variant which is rare and predicted to be deleterious by several algorithms. Currently Variant Ranker is restricted to human genome analysis (hg19 build).
Input/Output:The user is required to provide a valid e-mail, sample identifier and upload an input file in order to get started.
- Basic input to this tool is a simple text (.txt) file with Chr, Start, End, Ref, and Alt . allele columns (5 columns ONLY).
- Input also includes Variant Calling Format (VCF) format - general format of the output from most variant calling programs. Variant Ranker will take information and rank all the variants in the VCF file - it does not include sample/genotype information in its ranking algorithm and therefore ranking is restricted to single-sample VCF files and also biallelic variants.
- For multi-sample VCF file/(genotype filtering), users can use the CaseControl Filtering module in order to get an input list of variants that can be further ranked. See Example
- This tool is restricted to ranking of biallelic variants - when providing an input file, the INDELs get removed during the pre-processing stage and are provided separately for download. Please note multiallelic snps are decomposed into constituent SNPs.
| File format | ANNOVAR (please use .txt/.bed extensions & first 5 columns only)/ VCF file format |
| Input file size | 500 megabytes |
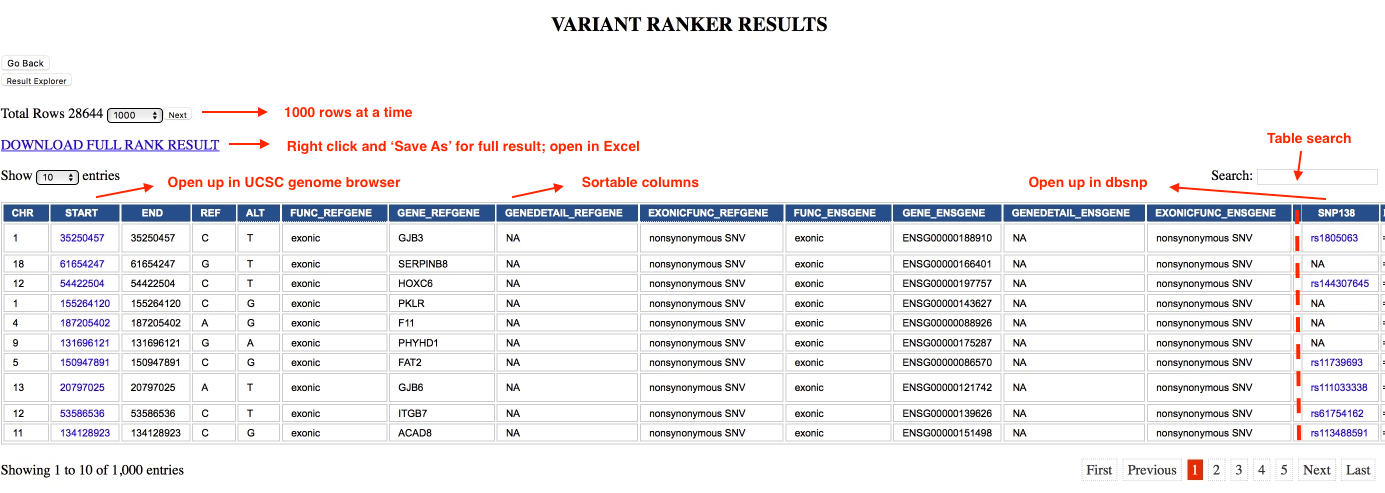
| VR Ranked table output | Sortable Table ordered by rank score; View 1000 variants at a time |
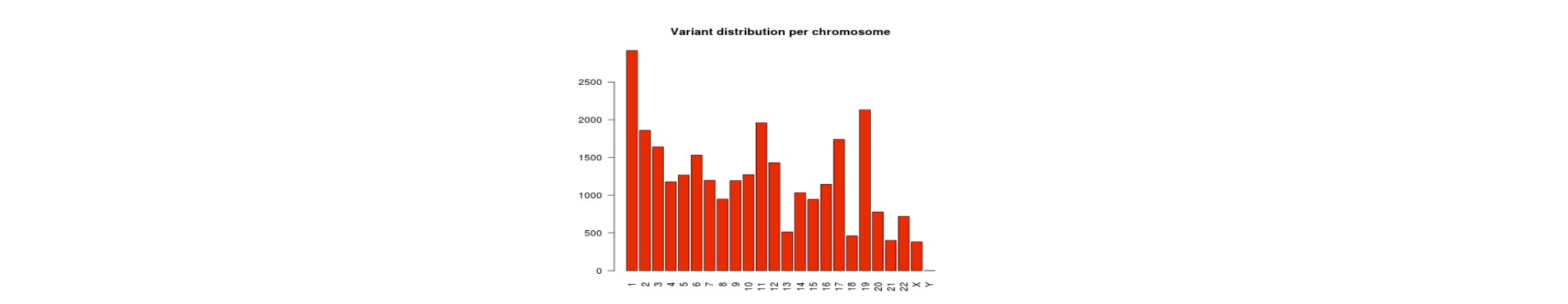
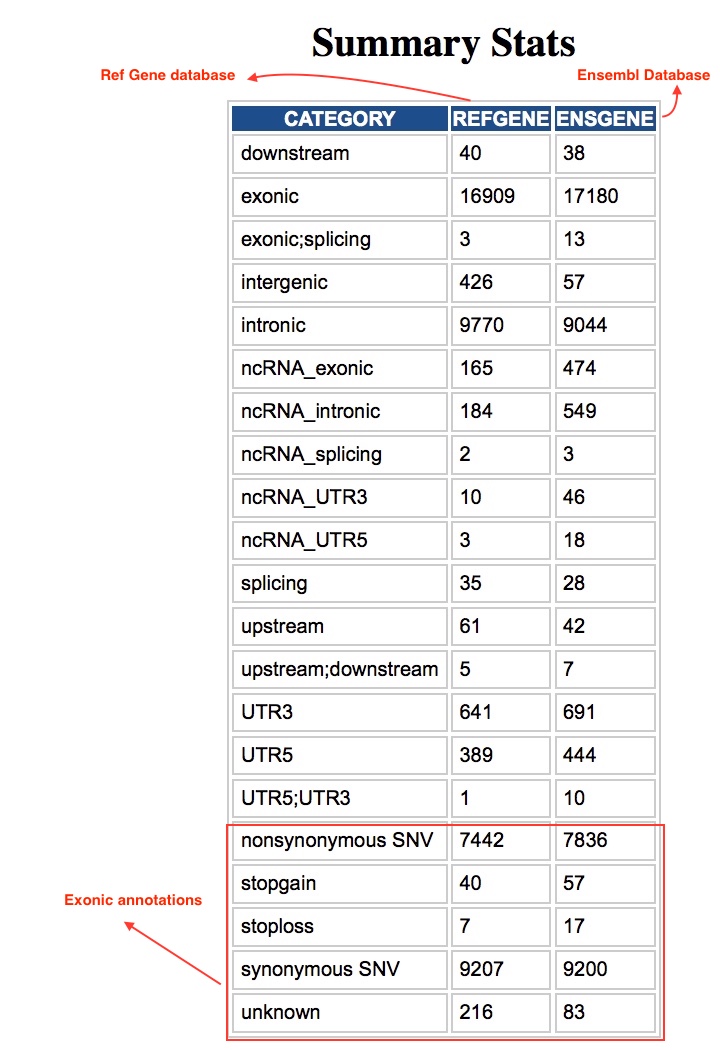
| Summary Statistics | Counts of variant categories |
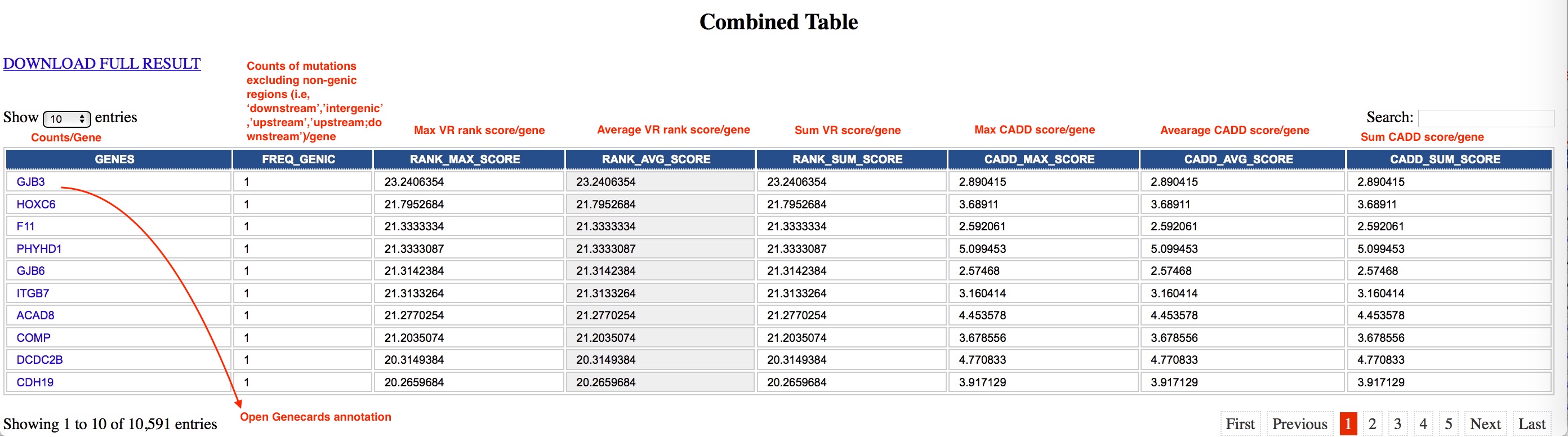
| Combined Table | Average counts per gene (see below details) |
| Example use | See Link |
The user will get a job submission number which on successful completion will receive an e-mail with downloadable ranked and annotated results. Job status of submitted job can be checked here. Results can be downloaded by right clicking and using Save As option on the DOWNLOAD FULL RESULT link. The .txt result can then be easily opened using Excel. Further filtering steps can be applied using the Result Explorer module.
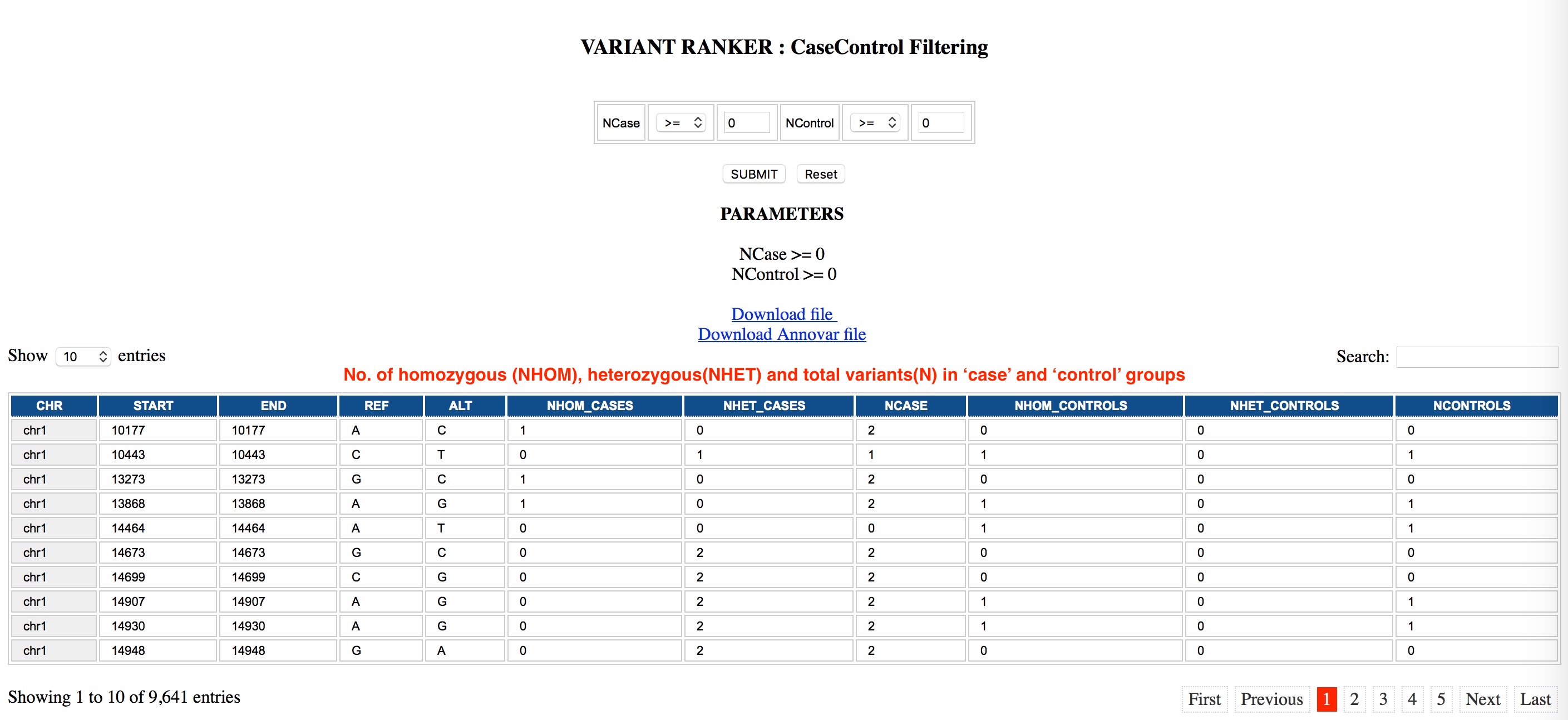
CaseControl Filtering:
Users can filter for variants between Cases/Controls, to obtain a list of variants that can be ranked using Variant Ranker. Users need to enter a SnpSift Case/Control command line string which is specified using '+','-','0' symbols; corresponds to the number of cases, controls or neutral (samples to ignore) samples respectively. Default filtering parameters are set to include all variants in the output. For faster post-processing we suggest using one of the parameters below (specifying an initial filtering criteria from the beginning).
INPUT: vcf or vcf.gz files (restricted to 500mb)PARAMETERS for filtering for variants in:
Job times were calculated based on the time between job submission and completion emails. The following should give the user an idea of processing times. Please note that the variable times would also depend on server work load i.e how many jobs aer working at the same time.
| Job ID | File type | Module | Total variants | Running Time |
| nonregistered-2017-06-16_12:42:53 | VCF | Variant Ranker | 28644 | 32 minutes |
| nonregistered-2017-06-16_12:50:32 | VCF | Variant Ranker | 30862 | 32 minutes |
| nonregistered-2017-06-15_18:22:08 | VCF | Variant Ranker | 155804 | 22 minutes |
| nonregistered-2017-06-15_18:14:13 | TXT | Variant Ranker | 155790 | 21 minutes |
| nonregistered-2017-06-16_02:06:04 | VCF | Variant Ranker | 1000055 | 4.5 hours |
| nonregistered-2017-06-16_13:38:12 | TXT | Variant Ranker | 1000055 | 5.47 hours |
| casectrl-nonregistered-2017-06-16_01:04:37 | VCF | CaseControl | 1000055 | 4 minutes |
Related publications:
In addition to real and synthetic validation datasets, we've applied our algorithm to targetted-resequencing data and family-exome sequencing data.
- Alexander J, Potamianou H, Xing J, Deng L, Karagiannidis I, Tsetsos F, Drineas P, Zsanett T, Rizzo R, Wolanczyk T, Farkas L, Nag y P, Szymanska U, Androutsos C, Tsironi V, Koumoula A, Barta C, Sandor P, Barr C, Tischfield J, Paschou P, Heiman G, Georgitsi M.(2016). Targeted re-sequencing approach of candidate genes implicates rare potentially functional variants in Tourette Syndrome etiology. Frontiers in Neuroscience.
- Alexander, J., Kalev, O., Mehrabian, S., Traykov, L., Raycheva, M., Kanakis, D., et al. (2016). Familial early-onset dementia with complex neuropathological phenotyp e and genomic background. Neurobiol. Aging.